The duty of each COVID-19 origin principle is to clarify a human outbreak in Wuhan, China, when the closest wild relations of SARS-CoV-2 are situated far-off, 1700 km to the South West.
In public, virologists have tended to say that the proximity of the outbreak to the Wuhan Institute of Virology, which uniquely specialises in accumulating, finding out, and enhancing, SARS-related coronaviruses, is a coincidence. As an alternative, they level to the close by Huanan seafood market because the possible spillover website, although it’s just like hundreds of others in China.
A Huanan market origin has formally been dismissed by the authorities in China. However, on February twenty fifth a preprint authored by George Gao, head of China’s CDC, and 38 different Chinese language virologists appeared that appears supposed to settle the difficulty (Gao et al., 2022).
The Gao article concludes, based mostly on a number of strains of proof, together with an absence of correlation between optimistic virus samples and stalls that bought animals, that the Huanan market was merely an amplifying occasion. These authors don’t specify how they thought the virus did first emerge, besides to notice that virus samples reported from different nations predate their Huanan market sampling by a number of months. This conclusion is in step with Chinese language authorities statements that SARS-CoV-2 got here from exterior China.
Simply sixteen hours later, on February twenty sixth, two preprints appeared concurrently that immediately contradict the Gao conclusions. The senior authors of those companion articles are an overlapping set of very high-profile virologists. None are from China.
One in all these preprints asserts, based mostly on floor swabs and different environmental samples discovered there, that the Huanan market was the “unambiguous epicenter” of the pandemic (Worobey et al., 2022). The second argues that SARS-CoV-2 emerged at the very least twice on the market (Pekar et al., 2022). In line with these latter authors, one zoonotic spillover created what are often called the lineage A SARS-CoV-2 viruses and a second spillover was the foundation of all lineage B SARS-CoV-2 viruses. These two spillovers, they are saying, decisively contradict a lab leak.
Many SARS-CoV-2 genomes discovered early within the outbreak are intermediate in sequence between the A and B lineages. These intermediates have beforehand been assumed to point a single spillover with one lineage evolving into the opposite (Morel et al., 2021; Pipes et al., 2021). Pekar et al. suggest as an alternative that such intermediates are all both artifactual (principally sequencing errors) or are in any other case irrelevant to the origin. Each papers are coy, nevertheless, about what kind of animal was concerned of their theorised spillovers.
These conflicting conclusions arrange an fascinating dynamic. Clearly, Chinese language virologists don’t assist the market speculation. Then again, the senior authors of Pekar et al. and Worobey et al. are outstanding Western virologists. Many, like Kristian Andersen, Robert Garry, Ed Holmes, and Andrew Rambaut, are vocal public supporters of a zoonotic origin, and really near Anthony Fauci the director of NIAID.
One facet of this dynamic is the East/West cut up. Clearly, the 2 factions usually are not co-operating. The opposite is a distinction of method. The Chinese language researchers assert what they suppose did not occur. In distinction, by formulating an specific speculation (aside from the host animal), the Western virologists have staked their credibility on a particular principle. The previous method is low danger; the latter is excessive danger since any particular principle is probably susceptible if new proof have been to return ahead that disproves it (as confirmed earlier instances would); however the reward has been ample media consideration of the “The Lab Leak is Useless” kind.
A major characteristic of this episode is the shut timing of the three papers. If extra proof was wanted of non-cooperation, it appears apparent that the Worobey/Pekar preprints have been an ambush. Their look was exactly timed to spike the headlines that the China CDC paper would doubtless have generated by ruling out a market origin.
Do Pekar and Worobey make their case?
Particularly if one contains the brand new Gao et al. proof, there are already highly effective causes to doubt each the market-zoonotic origin and a dual-spillover. These causes are largely glossed over by the Pekar and Worobey preprints so they’re price outlining briefly:
1) The market samples have been most likely taken late within the Wuhan outbreak.
The primary motive, and the only, is that the market samples have been taken between Jan 1st and March thirtieth, 2020. But plentiful proof, equivalent to contemporaneous newspaper reviews of an outbreak in Wuhan, implies that SARS-CoV-2 was circulating broadly in Wuhan and past by January 1st. Such proof makes it troublesome to agree that the market samples, so hotly mentioned, have any particular relevance to the supply of the pandemic virus itself.
For instance, in keeping with the WHO COVID origin investigation, there have been 174 COVID-19 hospitalisations in Wuhan by December thirty first 2019. Given the traditional delay between an infection and hospitalisation and the numerous fee at which COVID-19 offers asymptomatic and gentle instances, these hospitalisations doubtless represented solely the tip of a giant infectious outbreak in December.
Certainly, Ian Lipkin, an epidemiologist at Columbia College, advised an interviewer he knew of an outbreak in Wuhan by December fifteenth, 2019. Lipkin has subsequently confirmed this assertion. And in spring 2020, Peter Daszak, President of the EcoHealth Alliance, Marjorie Pollack, an epidemiologist who runs ProMED, and Public Well being Professor Lawrence Gostin made related statements to the LA Occasions. Additional again nonetheless, in keeping with ABC Information, US safety companies have been monitoring a pneumonia outbreak in Wuhan in November.
Early extensive unfold of the virus in Wuhan is evidenced too by an in depth case examine of a household from Guangdong who visited Wuhan between December twenty ninth and January 4th, 2020. 5 from a complete of six members of the family contracted COVID-19 whereas in Wuhan, with out them having visited any markets (Chan et al., 2020). Additional afield, a big physique of genome sequence and antibody proof suggests SARS-CoV-2 was in Europe and different nations within the fall of 2019, properly earlier than the Huanan market samples have been taken (reviewed in Canuti et al., 2022).
If there have been hundreds of instances within the metropolis of Wuhan by Jan 1st when the market was closed and 10,000 individuals per day often visited it, how do samples taken then (or later) represent credible proof for a market origin? Fairly doubtless, distributors and others on the market discovered to have COVID-19 infections have been simply typical for Wuhan in December 2019 (Courtier-Orgogoz and de Assis, 2022). Typical or not, the market samples have been collected too late to tell apart a market origin from another origin in or close to Wuhan.
2) Environmental samples collected on the market are of human origin and didn’t come from animals bought there.
The intention of the Chinese language CDC paper was to analyse the environmental samples (swabs from surfaces and so on.) that they took in and across the Huanan market after Jan 1st, 2020 (Gao et al., 2022). They concluded that the market was solely an amplifying occasion, partly as a result of SARS-CoV-2-positive samples have been related to stalls belonging to a number of kinds of distributors, together with these not promoting animals (the Worobey preprint argues there’s a correlation). Extra compelling, the CDC authors discovered that the samples collected from the market, which Pekar and Worobey declare are from contaminated animals, are admixed solely with human genetic materials and not with genetic materials from raccoon canines or different species probably bought on the market. The one affordable inference is that these optimistic samples didn’t derive from the faeces or urine or exhalations of a dwell non-human animal. Few outcomes would higher point out that virus-positive market samples derive from contaminated people versus different species.
3) Pekar and Worobey depend on round reasoning to establish root viruses.
The 2022 Pekar et al. preprint adapts the findings of a earlier publication (Pekar et al., 2021) to generate the novel speculation of a cut up phylogeny that traces SARS-CoV-2 again to 2 impartial spillovers, each occurring within the Huanan market. These two spillovers, they declare, are represented at the moment by what are often called lineage A and lineage B viruses, which differ by solely two mutations. Nonetheless, the phylogenetic strategies used for constructing evolutionary timber and thus figuring out the foundation virus in each Pekar papers are extremely problematic as a result of they’re susceptible to uneven and biased sampling and strange genetic phenomena, equivalent to superspreading occasions (Liu et al., 2020). One key bias related right here is that, for a lot of early COVID-19 instances, contact with the Huanan market was a diagnostic requirement (Liu et al., 2020). This may are inclined to orient phylogenies in the direction of the market. Additional, Pekar et al. use a clock-based algorithm that makes use of sampling dates to deduce the foundation virus. This methodology is designed to channel the selection of root virus in the direction of these genomes sampled earliest. If the market was the early focus of sampling, which it was, then the Pekar methodology of inferring the foundation is predicated on two impartial types of round reasoning. These biases have been additional amplified by the Pekar and Worobey authors, who themselves determined, based mostly on scant proof, which affected person instances counted in the direction of the dataset and typically what have been their illness onset dates. The impact of this intervention was so as to add but extra circularity into the choice course of for root viruses. To satisfactorily decide which viruses are closest to the true origin requires as an alternative a unique methodology, one that’s explicitly impartial of ascertainment biases and subjective decision-making (Liu et al., 2020).
4) Pekar et al., lack the proof for 2 spillover occasions.
A key assertion of the Pekar preprint is its proposal that extant lineage A and lineage B viruses signify the descendants of two impartial SARS-CoV-2 spillover occasions (Pekar et al., 2022). To succeed, this double spillover declare should clarify why quite a few genome sequences exist which are intermediate between lineage A and lineage B. To beat this problem, Pekar et al. suggest that such intermediates are all both artifacts from sequencing errors or irrelevant to the origin query for different causes. Sequencing errors are frequent sufficient, however Pekar et al. solely reveal them convincingly in a minority of cases. For instance, for many of their prompt sequence artifacts they depend on an unverifiable ‘private communication’ from a single scientist (L. Chen) in China. To make a case for the irrelevance of others they need to recommend, for instance, that two genomes sampled in February in Beijing are irrelevant–as if early sequences can’t have unfold elsewhere or continued. Finally, their daring suggestion that the phylogeny of SARS-CoV-2 is greatest defined by resolving it into two impartial spillovers may be very poorly supported by proof.
The Nice Virological Sport
Pekar and Worobey fail to make their case and therefore it’s tempting to dismiss their heavy reliance on weak knowledge, their non-parsimonious interpretations, their cherry-picking, and their round reasoning as easy shoddy science. However, in our expertise, this could be an error. Poor science by in any other case competent scientists, and on such a scale, often occurs for a motive. And, within the mild of the assembly, described by Katherine Eban of Self-importance Honest, between Jesse Bloom, Kristian Andersen, Anthony Fauci, Francis Collins, and others, which revolved round deleted sequences from early affected person samples, it appears extra clear than ever what that motive is.
Strange dangerous science principally happens for causes which are easy and mundane. Maybe a analysis subject is taken into account a scientific backwater to which second-rate researchers have gravitated, or a analysis thesis was badly supervised or not accomplished. If that’s the case, the outcomes are virtually positive to seem in a low-ranking peer-reviewed journal.
The opposite class of dangerous science suits a really completely different sample. It typically happens that the leaders who management science’s purse-strings decide to a principle or a significant programme that’s then contradicted by rising proof. If, for political or monetary causes, a coverage course correction is unavailable, a rationalisation in a outstanding scientific journal will likely be obligatory to supply, because the Vatican would possibly put it, “steerage for the devoted”.
Such publications usually have an unnecessarily giant variety of authors, who will principally be laboratory heads and different outstanding scientific leaders; the article will often seem in a journal with the very highest visibility, like Nature, Cell, or Science, and the errors such papers include (the rationalisations) are by no means errors–they’re purposeful and punctiliously calculated. A basic of this style, which we dissected intimately, was the NIH response to the failure of the human genome challenge to ship on its promise of explaining human noncommunicable illness, an issue that continues to be to this present day (Manolio et al., 2009).
Clearly, lab leak theories are a prime concern of the infectious illness group and coalescing opinion round a semi-plausible zoonotic speculation is the slightly apparent intent of Fauci’s NIAID. Granted, we don’t but know wherein journals Pekar et al., 2022 and Worobey et al., 2022 will seem, however their precursors (Pekar et al., 2021 and Worobey, 2021) have been each printed in Science. Earlier than that, the prototype for all future efforts was The proximal origin of SARS-CoV-2, printed in Nature Medication (Andersen et al., 2020).
Placing this all collectively, one can see that each Chinese language and Western virologists are pursuing the identical basic technique, step one of which is to ignore, discredit, deny, delete, destroy, or in any other case conceal, early sequences and samples (Bloom, 2021; Canuti et al., 2022). Erasing, or failing to gather, early info has the first impact of lessening the chance {that a} true origin will ever be retrieved. Secondarily, erasure additionally makes it simpler to power most well-liked conclusions on the info that stay. On the Chinese language facet, eradicating or failing to gather early proof of a Wuhan outbreak helps to put the primary documented look of the virus exterior of China solely. The Western aim has as an alternative been to power a market spillover conclusion by preferentially discrediting samples and instances that occurred previous to the market sampling and people with no hyperlinks to the market (Pekar et al., 2021 and Worobey, 2021).
These strategic goals can be unfeasible with out the circularity of the usual phylogenetic strategies mentioned above, that are fairly broadly understood by insiders (Liu et al., 2020; Kumar et al., 2021). Thus, selecting and selecting early samples lets clock-based phylogenetic strategies which are susceptible to sampling biases ship a foreordained root virus.
The key complication, seen to all after the publication of the conflicting Gao, Pekar, and Worobey preprints, is {that a} vital battle has arisen because of the divergent origin eventualities every group is attempting to suit the details to.
It’s a affordable inference from the above that the main virologists on either side, and who’re directing these efforts, strongly suspect (or know as a result of they’re sitting on the proof) that early knowledge wouldn’t exonerate virus analysis in Wuhan, in any other case the identical individuals can be attempting to find early samples with nice alacrity, which is clearly not taking place. And we are able to surmise it is a lab leak that’s being coated up since it’s the solely concern that each Chinese language and Western virologists might plausibly share.
Till just lately, the COVID-19 origin query was due to this fact set to devolve right into a easy two-way tug-of-war between the Gao membership within the East and the Fauci membership within the West. What none of them anticipated, nevertheless, was {that a} novel phylogenetic methodology would emerge able to discrediting their cautious calculus.
Mutational Order Evaluation
Just lately, a unique methodology has been utilized to the SARS-CoV-2 origin query (Kumar et al., 2021). This methodology is new to virology however it’s broadly utilized in most cancers analysis (e.g. Miura et al., 2018). Utilizing it, Kumar, Pond, and colleagues have been in a position to infer the existence of viral strains which are older (i.e. ancestors of) Wuhan-hu-1 (the usual SARS-CoV-2 reference genome) and the opposite market sequences by at the very least 3 mutations, which is so much.
Their progressive methodology is known as Mutational Order Evaluation (MOA). MOA is a crucial advance over normal approaches, not least as a result of it doesn’t depend on clocks (i.e. time) to orient (i.e. bias) the evolutionary timber it produces. Fairly, it makes use of genome sequence knowledge alone to infer the progenitor virus. Thus, MOA can be utilized to undo identified biases, equivalent to clocks, different sampling confounders, and even systematic pattern destruction.
To know the key distinction between MOA and the phylogenetic methodology utilized by Pekar et al., think about a theoretical particular person in Wuhan in late 2019 who caught a really early case of SARS-CoV-2 and who then flew to a distant nation. As soon as there, they seeded a minor outbreak that lasted just some weeks or months (not in contrast to the Guangdong household famous earlier). If any genome from a later case on this outbreak have been by probability sequenced, this info can be very precious. It might be a uncommon instance of a root virus genome (or very near it). From this hypothetical instance we are able to see that even sequences that happen late in an epidemic, or removed from a presumed geographic origin, can, in precept, protect important details about that origin.
Ordinarily, phylogenetic evaluation of the origin (together with Pekar et al., 2021 and 2022 and Bloom, 2021) tends to focus, typically solely, on early sequences and people discovered native to the outbreak origin. Genomes thought of of inconceivable relevance to the origin query are ignored. For example, Pekar et al. 2022 carried out their evaluation on 787 genomes, with a closing date of February 14th, 2020. MOA, nevertheless, can use each out there genome sequence to construct up an image of viral relatedness, with out discriminating. If any virus genome within the knowledge set constitutes an obvious lacking hyperlink between two viruses it is going to be inserted as a part of the mutational order that’s constructed up. As a result of MOA makes use of a really giant knowledge set, particularly to seize idiosyncratic occasions such because the one theorised above, it may possibly construct a much more correct, much more detailed, and way more statistically sturdy evolutionary tree than standard approaches. Better of all, it does so with out introducing any biases of its personal.
For these causes, MOA is clearly a superior methodology. Its worth is particularly nice for inferring outbreak origins in instances the place, like SARS-CoV-2, early sequences are scarce and their assortment is topic to sampling biases.
MOA contradicts the Pekar and Worobey twin spillover market principle
1) MOA identifies only one root virus.
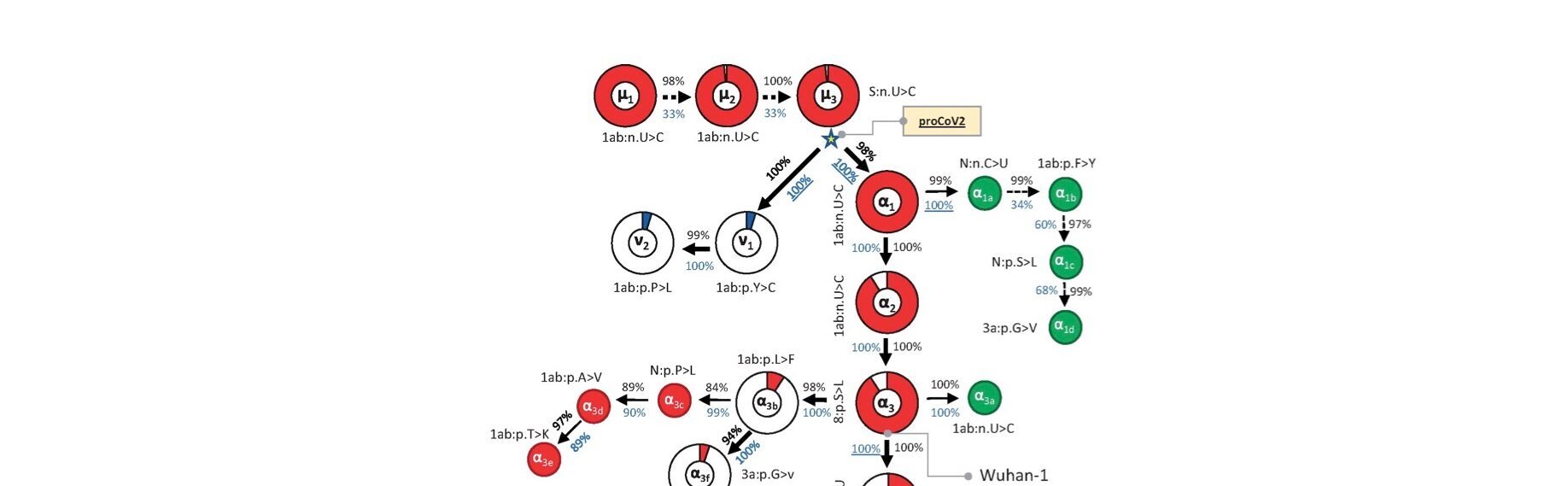
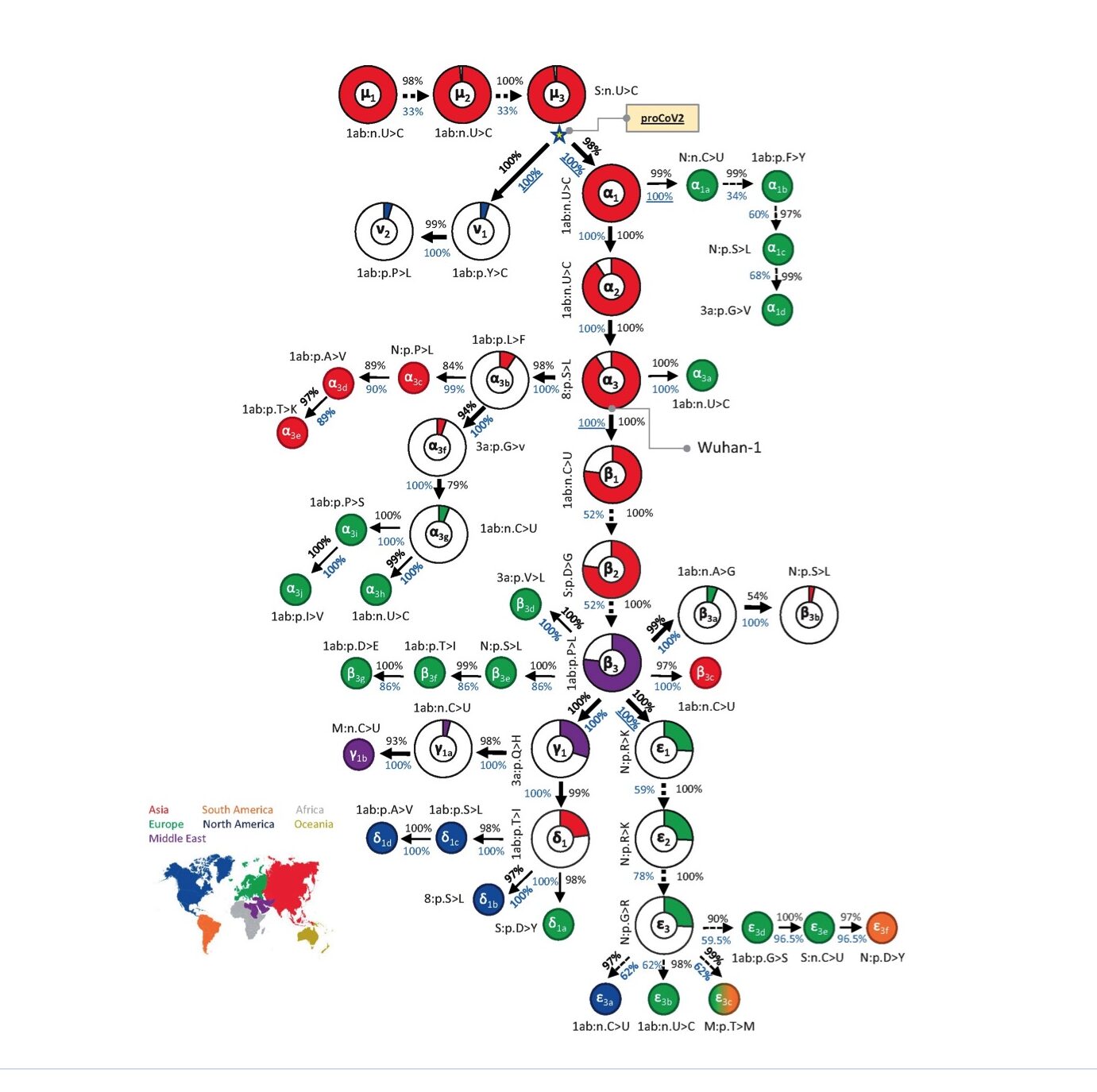
From virtually 176,000 full-length genomes, MOA was in a position to decipher a mutational order for the origin viruses, with very excessive statistical confidence (Kumar et al., 2021). Their outcomes are summarised in Fig. 1 under (taken from Fig. 2 of Kumar et al., 2021).
Fig. 1 The phylogeny of the early pandemic, in keeping with Kumar et al., 2021
Total, three of its findings strongly contradict the twin spillover market principle:
1) As is clear from Fig. 1, in contrast to Pekar et al. (2022), MOA identifies a single root virus (μ1, prime left, is its first mutant). A single root virus means the pandemic started with just one preliminary spillover. A single spillover occasion is a vital statement as a result of it strongly implies a lab leak (since scientists are inclined to work with pure cultures); whereas equal proof for a number of and/or genetically numerous spillovers would have implied a pure supply. MOA additionally exhibits that each one lineage B viruses are descended from one lineage A virus.
2) The foundation virus recognized by MOA predates all the foundation viruses recognized by Pekar and Worobey.
The virus recognized by MOA as the foundation is separated by a number of mutations from any of the viral genomes discovered on the Huanan market and from Wuhan-hu-1. This infers that each one identified market samples are properly downstream of affected person zero. Due to this fact too, as was additionally concluded by Gao et al., the market samples signify an amplifying occasion, at most.
Evolution takes time to happen. As a result of a number of mutations collected earlier than the primary confirmed instances, Kumar et al. calculated that the preliminary spillover (presumably in Wuhan) was round late October/early November, i.e. a number of months earlier than the Wuhan market was first sampled (on Jan 1st, 2020). This conclusion is therefore per the presence of far-flung SARS-CoV-2 genomes in Europe and elsewhere within the fall of 2019. Additionally it is per extensive unfold of the virus in Wuhan earlier than the market samples have been collected.
Very just lately, Kumar and colleagues launched an additional preprint (Caraballo-Ortiz et al., 2022). It makes use of much more knowledge (1 million genomes) and an improved methodology, which they name TopHap, to make their findings nonetheless extra sturdy. The addition of many extra genome sequences, together with some very near the foundation, affirms the unique conclusions of a single root virus and that lineage B developed from lineage A. It additionally allowed them to maneuver again the foundation virus by one additional mutation and this pushes again even additional in time the anticipated date of SARS-CoV-2 emergence–into September, 2019.
This September spillover date additional contradicts Pekar and Worobey’s date. Nonetheless, it agrees properly with a broad set of different phylogenetic analyses and makes the farflung virus findings in Europe and elsewhere extra believable nonetheless (Mostefai et al., 2021; Schrago and Barzilai, 2021; Track et al., 2021; Xia, 2021).
3) Analysing solely viruses present in China decided the outcomes of the Pekar articles. Of their phylogenetic analyses, each Pekar papers are noteworthy for choosing slender geographical and temporal cut-offs for his or her knowledge units. Although these decisions appear, at first sight, affordable sufficient, as Kumar et al. level out for Pekar et al., 2021, the alternatives decide their final conclusions. The excluding of sequences obtained exterior China (and likewise these sampled greater than 4 months after December) prevented these authors from together with the key early department that started with v1 (see Fig. 1). The v lineage was first sampled within the US however is required for the proper number of a root virus (see additionally Morel et al., 2021). Finally, the entire exclusion of the v lineage due to make use of of slender cut-offs is a key motive why the 2 approaches reached divergent conclusions (Kumar et al., 2021). In brief, the scientific contribution of Pekar et al. (2021 and 2022) is to deploy slender home windows for knowledge acquisition to perform the phylogenetic equal of p-hacking.
If Pekar and Worobey are incorrect, are Gao et al. proper?
The MOA methodology is just considerably kinder to the Gao et al. origin conclusions.
Though MOA helps the market being a secondary website, it contradicts the thought of a virus origin exterior China. Gao at al. level at proof for very early instances exterior of China and suggest that one in every of these was the final word supply. However, it’s clear from the Kumar et al. evaluation that Wuhan and China are the place the genetic range across the root happens. In different phrases, the virus is unlikely to have emerged in Italy (or elsewhere) in late 2019 and seeded Wuhan. Way more most likely, it spilled over in Wuhan and seeded Italy.
The standing of a zoonotic origin within the mild of MOA
We described above seven main flaws within the speculation of a double market spillover. Most of those criticisms apply to any hypothesised spillover situation on the Huanan market, however it’s notable that the MOA findings of Kumar, Pond, and colleagues advance the critique very considerably. Their two papers thus signify vital landmarks within the examine of the origin of COVID-19 (Kumar et al., 2021; Caraballo-Ortiz et al., 2022).
The important thing factors are price recalling: The in depth proof of an epidemic in Wuhan lengthy earlier than the Huanan market samples have been taken; that the market samples containing SARS-CoV-2 have been blended with human RNA and not with animal RNA; the round reasoning of Pekar et al.’s phylogenetic methodology; the shortage of assist for dismissing viruses intermediate between lineages A and B; the methodological superiority of the MOA methodology, which identifies a single and considerably earlier root; and, final, the dependence of the Pekar evolutionary tree on ignoring SARS-CoV-2 sequences harvested both late or exterior of China.
There may be additionally the broader context to think about. In case it wants reiterating, there’s nonetheless no proof for wild or farmed animals being contaminated with SARS-CoV-2 in China, both earlier than, throughout, or for the reason that pandemic broke out. Furthermore, zoonotic theorists have been very reluctant to specify a transparent candidate animal species as an intermediate host. This appears to be as a result of it’s onerous to assemble an excellent case for any of them. Third, there’s the unremarkable nature, by Chinese language requirements, of the Huanan market. Why Wuhan? It’s a query nonetheless unanswered by pure zoonotic theories.
In brief, it’s unreasonable to assert that the Huanan market was the “unambiguous epicentre” of the pandemic (Worobey et al., 2022). Such certitude is scientifically unwarranted and solely serves to provide the looks of a false flag operation.
This impression is strengthened since neither Gao, Pekar, nor Worobey, ever focus on the existence of the Wuhan Institute of Virology (WIV) and the plain various speculation that they know goes together with it. The WIV is just some miles from the market and, in keeping with its US funders and the individuals who work there, it specialises within the assortment and examine and enhancement of SARS-related coronaviruses (Latinne et al., 2020). For many years, a significant aim of its analysis has been to establish or create ones primed for human spillover (e.g. Li et al., 2019).
MOA, a goldmine of pandemic origin info
The 2 MOA/TopHap papers present by far the strongest candidate but for a root virus they usually element its subsequent evolution (Caraballo-Ortiz et al., 2022; Kumar et al., 2021). Consequently, they’ve much more to inform us as a result of exact rooting is invaluable for understanding extra key facets of any virus origin.
One key query to ask any zoonotic origin speculation is whether or not the foundation virus might infect putative intermediate hosts, such because the raccoon canine prompt by Pekar and Worobey. Answering this requires understanding exactly what these early strains have been. Such exams will be extremely deceptive if carried out on virus strains from a lot later within the pandemic for the reason that variants of SARS-CoV-2 have distinct mammalian host ranges (Montaguteli et al., 2021; Gu et al., 2020). Specifically, it ought to hassle origin theorists that the one proof for SARS-CoV-2 infecting raccoon canines comes from a later (D614G) pressure (Freuling et al., 2021).
A second advantage of correct rooting might be essentially the most vital of all. It follows from the truth that root viruses can present what, if any, have been the preliminary adaptation steps of SARS-CoV-2 to people.
For instance, the MOA phylogeny signifies (although this may’t be seen in Fig. 1) that even the very early virus strains that predated the market samples, continued unchanged lengthy into the pandemic. It is a crucial statement. It exhibits that, however later enhancements in its health, SARS-CoV-2 was very properly tailored to people by September or October and, as far as we are able to inform, from the very begin.
Corroborating this, the very earliest strains differ solely by synonymous mutations (see e.g. mutants μ1-3 in Fig. 1). Synonymous (versus nonsynonymous) mutations are nucleotide adjustments solely; that’s, they don’t alter the amino acid sequence and they also often have zero impact on the health of the virus.
(Word: For every mutant, Fig.1 exhibits both nucleotide adjustments (>) or amino acid adjustments (>). Nucleotides are represented by the letters A, C, G, or U and adjustments to those signify synonymous mutations; different letters signify the usual amino acid notation system and these letters thus point out nonsynonymous mutations.)
From the synonymous nature of the earliest mutations, the authors concluded that these earliest viruses:
“already possessed the repertoire of protein sequences wanted to contaminate, unfold, and persist within the international human inhabitants” (Kumar et al., 2021).
Furthermore, many rapid descendants of those viruses typically additionally contained solely synonymous mutations and but these strains too typically grew to become ample. Certainly, such barely altered genomes have been discovered “on each sampled continent” (Kumar et al., 2021).
Once more, this means the identical conclusion. Whichever manner one appears on the proof, it’s evident that even the very earliest viruses weren’t solely extremely tailored to people however in a position, unaltered, to trigger a pandemic.
It is a tremendously informative consequence. The query of whether or not SARS-CoV-2 was extremely tailored to people (and thus presumably preadapted) has been in sizzling dispute virtually from the start of the pandemic (Zhan et al., 2020). However the MOA phylogeny supplies by far essentially the most decisive proof but that certainly it was.
Studying into their phylogeny a little bit additional offers much more assist for preadaptation. It was not till the eighth mutation did one come up (β2 in Fig. 1) that has subsequently been proven to extend the health of the virus in people (Dearlove et al., 2020; van Dorp et al., 2020). β2 is the well-known D614G mutation, first recognized in Wuhan late January. By this time the pandemic was properly underway.
The one different mutation in Fig.1 that grew to become ample throughout the pandemic was its rapid predecessor, β1. β1 is a synonymous mutation (once more) that most likely hitch-hiked on D614G (Dearlove et al., 2020).
The importance of this sample is to point that, although they gave amino acid adjustments, even the nonsynonumous mutations clustered on the root have been selectively impartial. That’s, they too arose at random and never as a result of they conferred any benefit on the virus. This once more strongly reinforces the concept the virus was below little stress to adapt throughout its early unfold in Wuhan.
It’s not in any respect regular for a virus to enter a brand new host inhabitants with out additionally evolving quickly to adapt to it. Thus the primary SARS coronavirus (SARS-CoV) acquired amino acid adjustments throughout its early unfold in people (Zhan et al., 2020). The choice norm is for the virus to fail to adapt to its new host in any respect, equivalent to has occurred thus far with each one of many many introductions into people of the coronavirus MERS (Dudas et al., 2018).
Conceding that SARS-CoV-2 was preadapted, some have argued that SARS-CoV-2 is a ‘generalist’ virus (Frutos et al., 2020). There appears to be little or no proof for this. SARS-CoV-2 doesn’t infect most mammal species (Kock and Caceres-Escobar, 2022). It may be actively transmitted by far fewer species nonetheless, and, when these few do transmit the virus, adaptive mutations happen (Gu et al., 2021; Sawatzki et al., 2021; Tan et al., 2022). In brief, SARS-CoV-2 has not confirmed preadapted to any mammalian host species examined thus far (besides people) and so it bears not one of the hallmarks of a generalist virus.
Preadaptation of SARS-CoV-2 implies a lab leak. Nevertheless it additionally implies a leak of a particular type of virus; one that’s not merely tailored to human cells however to transmission between entire, intact, people. Just one principle of how SARS-CoV-2 arose suits this description. It’s the Mojiang Miners Passage principle.
The place now for origin analysis?
Main establishments, primarily the EcoHealth Alliance, the NIH, and the WIV, maintain giant troves of knowledge that would show or disprove a lab leak. All assert their transparency and accountability, however, in apply, every has denied quite a few requests for paperwork and different knowledge. If one solely listened to their phrases, one would suppose they needed to search out the origins of COVID-19, however their actions converse louder. Progress will due to this fact doubtless depend upon impartial initiatives.
When Jesse Bloom ingeniously retrieved beforehand deleted early SARS-CoV-2 sequences from the cloud and thereby recovered a novel early virus pressure (Bloom, 2021), virologist Rasmus Nielsen said that this info was:
“a very powerful knowledge that we have now acquired relating to the origins of COVID-19 for greater than a 12 months”.
Nielsen was confirmed right since Bloom’s discovery has been essential to the improved rooting of Kumar and colleagues (Caraballo-Ortiz, et al., 2022).
The distinctive analysis worth of those early sequences signifies additionally the suitable scientific benchmark for judging those that erase or withold such proof. Historical past is filled with ironies, however not many exceed the spectacle of outstanding scientists, whose careers are publicly funded on the promise of figuring out causes of infectious illnesses, not wanting others to know the reason for the COVID-19 pandemic. Why, in any case, did public establishments like NIH, NIAID, USAID, DOD, who’ve showered cash on the EcoHealth Alliance, supposedly to stop pandemics, not fund widespread searches for early SARS-CoV-2 infections on the very begin of the pandemic?
Given this blockade, even at this late date, most likely the only and most helpful strategy to advance the scientific seek for the origin of SARS-CoV-2 can be to find and analyse extra samples from early within the outbreak, from any nation, regardless (Basavaraju et al., 2021; Montomoli et al; 2021; Canuti et al., 2022). Preliminary analysis suggests a plethora of appropriate scientific and environmental samples exist in civilian and likewise army pattern collections and databases, and that searches for early sequences are more likely to be fruitful (Canuti et al. 2022, Paixao et al. 2022, Althoff et al. 2021; Chapleau et al. 2021; Lednicky et al. 2021; Basavaraju et al. 2021; Chen et al. 2020).
The opposite obligatory benchmark is an moral one. Dr Tedros of the WHO has known as investigating the origin of the pandemic “an ethical obligation“. We’d go additional. Though there isn’t any regulation in opposition to obstructing origin analysis, it ought to nonetheless be thought of against the law in opposition to all humanity to make it extra doubtless that an occasion that resulted in thousands and thousands of deaths and untold distress will recur as a result of we by no means discovered its trigger (Relman, 2020).
References
Andersen, Ok. G., Rambaut, A., Lipkin, W. I., Holmes, E. C., & Garry, R. F. (2020). The proximal origin of SARS-CoV-2. Nature drugs, 26(4), 450-452.Bloom, J. D. (2021). Restoration of deleted deep sequencing knowledge sheds extra mild on the early Wuhan SARS-CoV-2 epidemic. Molecular biology and evolution, 38(12), 5211-5224.
Althoff, Ok. N., Schlueter, D. J., Anton-Culver, H., Cherry, J., Denny, J. C., Thomsen, I., … & Schully, S. D. (2022). Antibodies to extreme acute respiratory syndrome coronavirus 2 (SARS-CoV-2) in All of Us Analysis Program individuals, 2 January to 18 March 2020. Medical Infectious Ailments, 74(4), 584-590.
Basavaraju, S. V., Patton, M. E., Grimm, Ok., Rasheed, M. A. U., Lester, S., Mills, L., … & Stramer, S. L. (2021). Serologic testing of US blood donations to establish extreme acute respiratory syndrome coronavirus 2 (SARS-CoV-2)–reactive antibodies: December 2019–January 2020. Medical Infectious Ailments, 72(12), e1004-e1009.
Caraballo-Ortiz, M. et al., (2022) TopHap: Fast inference of key phylogenetic buildings from frequent haplotypes in giant genome collections with restricted range. Bioinformatics, btac186
Chapleau, R. R., Christian, M., Connors, B., Premo, C., Chao, T. C., Rodriguez, J., … & Starr, C. (2021). Early Identification of SARS-CoV-2 Emergence within the DoD by way of Retrospective Evaluation of 2019-2020 Higher Respiratory Sickness Samples. medRxiv.
Courtier-Orgogozo, V., & de Ribera, F. A. (2022). SARS-CoV-2 an infection on the Huanan seafood market. Zenodo.
Chen, C., Li, J., Di, L., Jing, Q., Du, P., Track, C., … & Wang, J. (2020). MINERVA: a facile technique for SARS-CoV-2 whole-genome deep sequencing of scientific samples. Molecular cell, 80(6), 1123-1134.
Dearlove, B., Lewitus, E., Bai, H., Li, Y., Reeves, D. B., Joyce, M. G., … & Rolland, M. (2020). A SARS-CoV-2 vaccine candidate would doubtless match all at present circulating variants. Proceedings of the Nationwide Academy of Sciences, 117(38), 23652-23662.
Dudas, G., Carvalho, L. M., Rambaut, A., & Bedford, T. (2018). MERS-CoV spillover on the camel-human interface. elife, 7, e31257.
Frutos, R., Serra-Cobo, J., Chen, T., & Devaux, C. A. (2020). COVID-19: Time to exonerate the pangolin from the transmission of SARS-CoV-2 to people. An infection, Genetics and Evolution, 84, 104493.
Gao, G., Liu, W., Liu, P., Lei, W., Jia, Z., He, X., … & Wu, G. (2022). Surveillance of SARS-CoV-2 within the surroundings and animal samples of the Huanan Seafood Market.
Gu, H., Chen, Q., Yang, G., He, L., Fan, H., Deng, Y. Q., … & Zhou, Y. (2020). Adaptation of SARS-CoV-2 in BALB/c mice for testing vaccine efficacy. Science, 369(6511), 1603-1607.
Kock, R., and Caceres-Escobar, H. (2022) State of affairs evaluation on the roles and dangers of wildlife within the emergence of human infectious illnesses. IUCN
Kumar, S., Tao, Q., Weaver, S., Sanderford, M., Caraballo-Ortiz, M. A., Sharma, S., … & Miura, S. (2021). An evolutionary portrait of the progenitor SARS-CoV-2 and its dominant offshoots in COVID-19 pandemic. Molecular Biology and Evolution, 38(8), 3046-3059.
Latinne, A., Hu, B., Olival, Ok. J., Zhu, G., Zhang, L., Li, H., … & Daszak, P. (2020). Origin and cross-species transmission of bat coronaviruses in China. Nature Communications, 11(1), 1-15.
Li, H., Mendelsohn, E., Zong, C., Zhang, W., Hagan, E., Wang, N., … & Daszak, P. (2019). Human-animal interactions and bat coronavirus spillover potential amongst rural residents in Southern China. Biosafety and well being, 1(02), 84-90.
Lednicky, J., Salemi, M., Subramaniam, Ok., Waltzek, T. B., Sabo-Attwood, T., Loeb, J. C., … & Morris Jr, J. G. (2021). Earliest detection so far of SARS-CoV-2 in Florida: Identification along with influenza virus on the principle entry door of a college constructing, February 2020. Plos one, 16(1), e0245352.
Liu, Q., Zhao, S., Shi, C. M., Track, S., Zhu, S., Su, Y., … & Chen, H. (2020). Inhabitants genetics of SARS-CoV-2: disentangling results of sampling bias and an infection clusters. Genomics, proteomics & bioinformatics, 18(6), 640-647.
Manolio, T. A., Collins, F. S., Cox, N. J., Goldstein, D. B., Hindorff, L. A., Hunter, D. J., … & Visscher, P. M. (2009). Discovering the lacking heritability of advanced illnesses. Nature, 461(7265), 747-753.
Miura, S., Huuki, L. A., Buturla, T., Vu, T., Gomez, Ok., & Kumar, S. (2018). Computational enhancement of single-cell sequences for inferring tumor evolution. Bioinformatics, 34(17), i917-i926.
Montagutelli, X., Prot, M., Levillayer, L., Salazar, E. B., Jouvion, G., Conquet, L., … & Simon-Loriere, E. (2021). The B1. 351 and P. 1 variants prolong SARS-CoV-2 host vary to mice. BioRxiv.
Morel, B., Barbera, P., Czech, L., Bettisworth, B., Hübner, L., Lutteropp, S., … & Stamatakis, A. (2021). Phylogenetic evaluation of SARS-CoV-2 knowledge is troublesome. Molecular biology and evolution, 38(5), 1777-1791.
Mostefai, F., Gamache, I., N’Guessan, A., Pelletier, J., Huang, J., Murall, C. L., … & Hussin, J. (2022). Inhabitants genomics approaches for genetic characterization of SARS-CoV-2 lineages. Frontiers in drugs, 207.
Paixao, J., Galangue, M., Gaston, C., Carralero, R., Lino, C., Júlio, G., … & Francisco, N. M. (2022). Early Proof of Circulating SARS-CoV-2 in Unvaccinated and Vaccinated Measles Sufferers, September 2019–February 2020. An infection and Drug Resistance, 15, 533.
Pekar, J., Worobey, M., Moshiri, N., Scheffler, Ok., & Wertheim, J. O. (2021). Timing the SARS-CoV-2 index case in Hubei province. Science, 372(6540), 412-417.
Pekar, J., et al., (2022) SARS-CoV-2 emergence very doubtless resulted from at the very least two zoonotic occasions. Zenodo.
Pipes, L., Wang, H., Huelsenbeck, J. P., & Nielsen, R. (2021). Assessing uncertainty within the rooting of the SARS-CoV-2 phylogeny. Molecular biology and evolution, 38(4), 1537-1543.
Relman, D. A. (2020). Opinion: To cease the following pandemic, we have to unravel the origins of COVID-19. Proceedings of the Nationwide Academy of Sciences, 117(47), 29246-29248.
Schrago, C. G., & Barzilai, L. P. (2021). Challenges in estimating virus divergence instances briefly epidemic timescales with particular reference to the evolution of SARS-CoV-2 pandemic. Genetics and molecular biology, 44.
Track, N., Cui, G. L., & Zeng, Q. L. (2021). Genomic epidemiology of SARS-CoV-2 from Mainland China. Frontiers in microbiology, 12, 1211.
van Dorp, L., Richard, D., Tan, C., Shaw, L. P., Acman, M., & Balloux, F. (2020). No proof for elevated transmissibility from recurrent mutations in SARS-CoV-2. Nature communications, 11(1), 1-8.
Tan, C., Lam, S. D., Richard, D., Owen, C., Berchtold, D., Orengo, C., … & Balloux, F. (2022). Transmission of SARS-CoV-2 from people to animals and potential host adaptation.
Worobey et al. (2022) The Huanan market was the epicenter of SARS-CoV-2 emergence. Zenodo.
Xia, X. (2021). Relationship the Widespread Ancestor from an NCBI Tree of 83688 Excessive-High quality and Full-Size SARS-CoV-2 Genomes. Viruses, 13(9), 1790.
Zhan, S. H., Deverman, B. E., & Chan, Y. A. (2020). SARS-CoV-2 is properly tailored for people. What does this imply for re-emergence?. BioRxiv.
If this text was helpful to you please think about sharing it along with your networks.